Welcome to CoSMoS
Computations in Structural, Molecular, and Systems biology

DeepPROTECTNeo is a comprehensive web server designed to streamline neoantigen discovery for personalized cancer vaccines. By processing Whole Exome Sequencing (WES) or Whole Genome Sequencing (WGS) data, it integrates HLA typing, TCR profiling, peptide-MHC binding prediction, and TCR-epitope interaction assessment into a single workflow. Powered by advanced deep learning, including context-aware transformers, it achieves unparalleled precision in neoepitope discovery and supports multi-epitope vaccine design. With extensive validation on clinical datasets like TESLA, DeepPROTECTNeo offers researchers and clinicians an innovative tool to advance cancer immunotherapy.
Under review
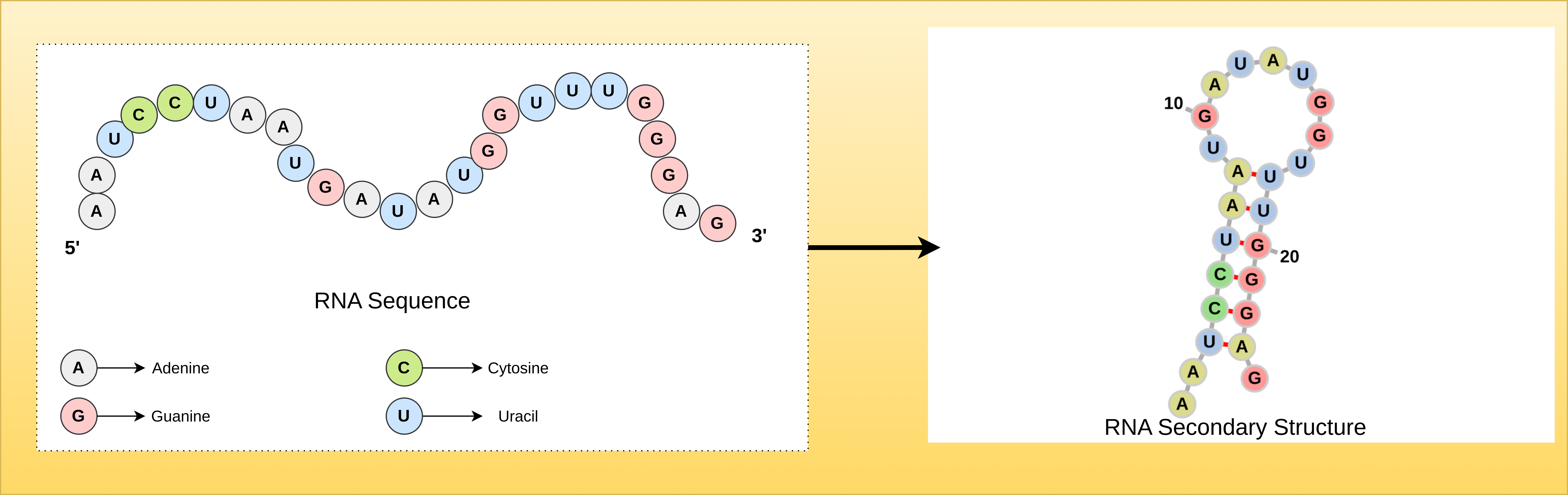
rpcFold can efficiently handle RNA with any length and outperforms the existing models on both within-family and cross-family RNA datasets.
Under review
Capping is a post-transcriptional modification of RNA, where a cap is formed at the 5’ end of an RNA transcript. Cap Analysis Gene Expression(CAGE) is a protocol designed to read and measure the expression of 5’ associated caps of RNAs. CAGE is based on “cap-trapping”, a method in which the 5’ cDNA is pulled down following the biotenylation of the 7-methylguanosine cap of the corresponding transcript. CAGE Tags refer to the capping regions pulled by the cap trapping method. CAGETag is a transformer-based web server that predicts the CAGE region.
Under review
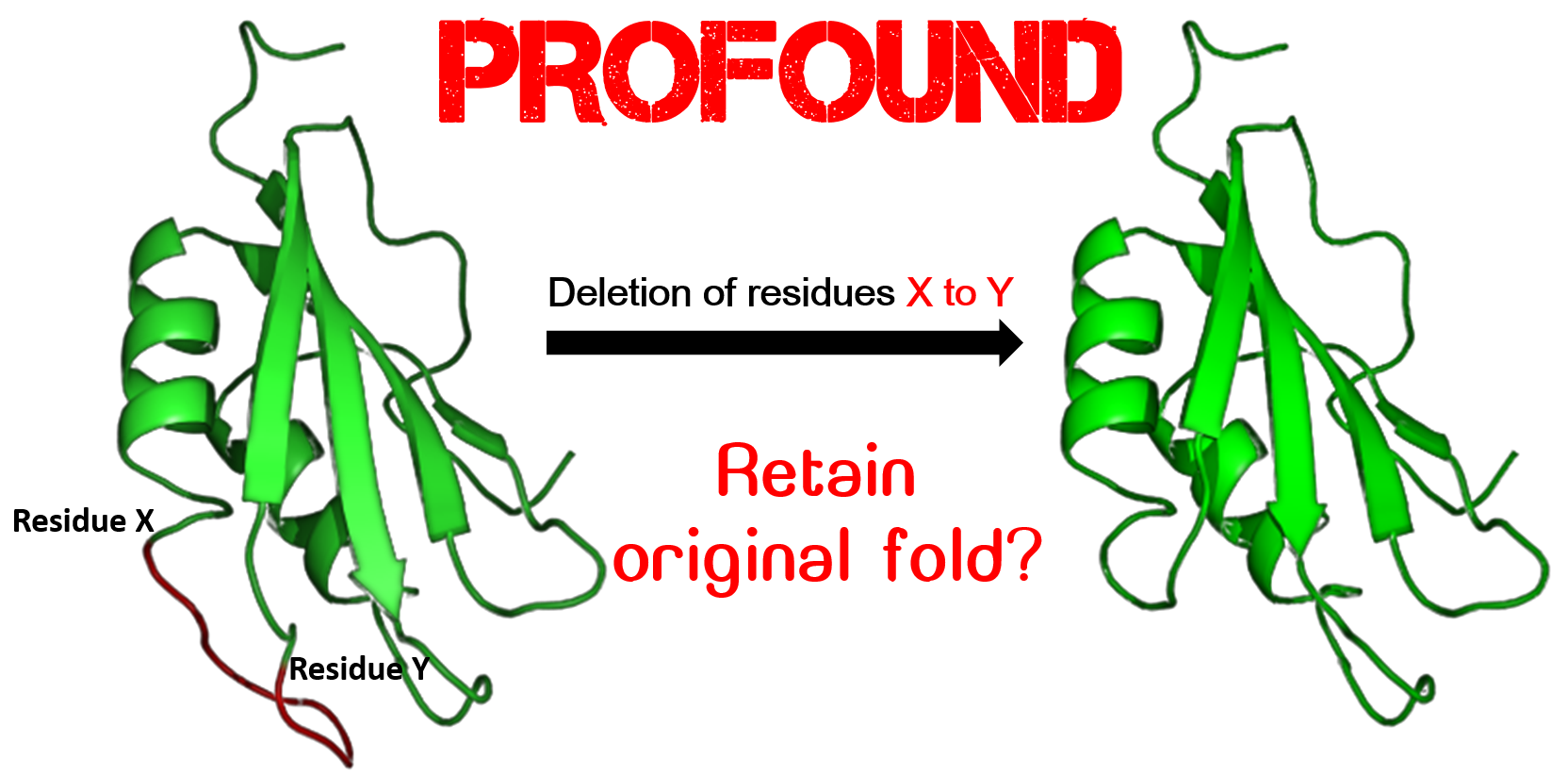
PROFOUND uses positive-unlabeled classifiers utilizing fold level attributes, environment-specific properties, and deletion site-specific properties to predict the change in foldability owing to contiguous multi-point deletion (MPD) of amino acids in protein structures. PROFOUND is a first of its kind prediction system that reports a recall of 82.2% (86.6%) and a fall out rate of 14.2% (20.6%) corresponding to MPDs in the loop (non-loop) region. J Chem Inf Model 60(12):6679-6690
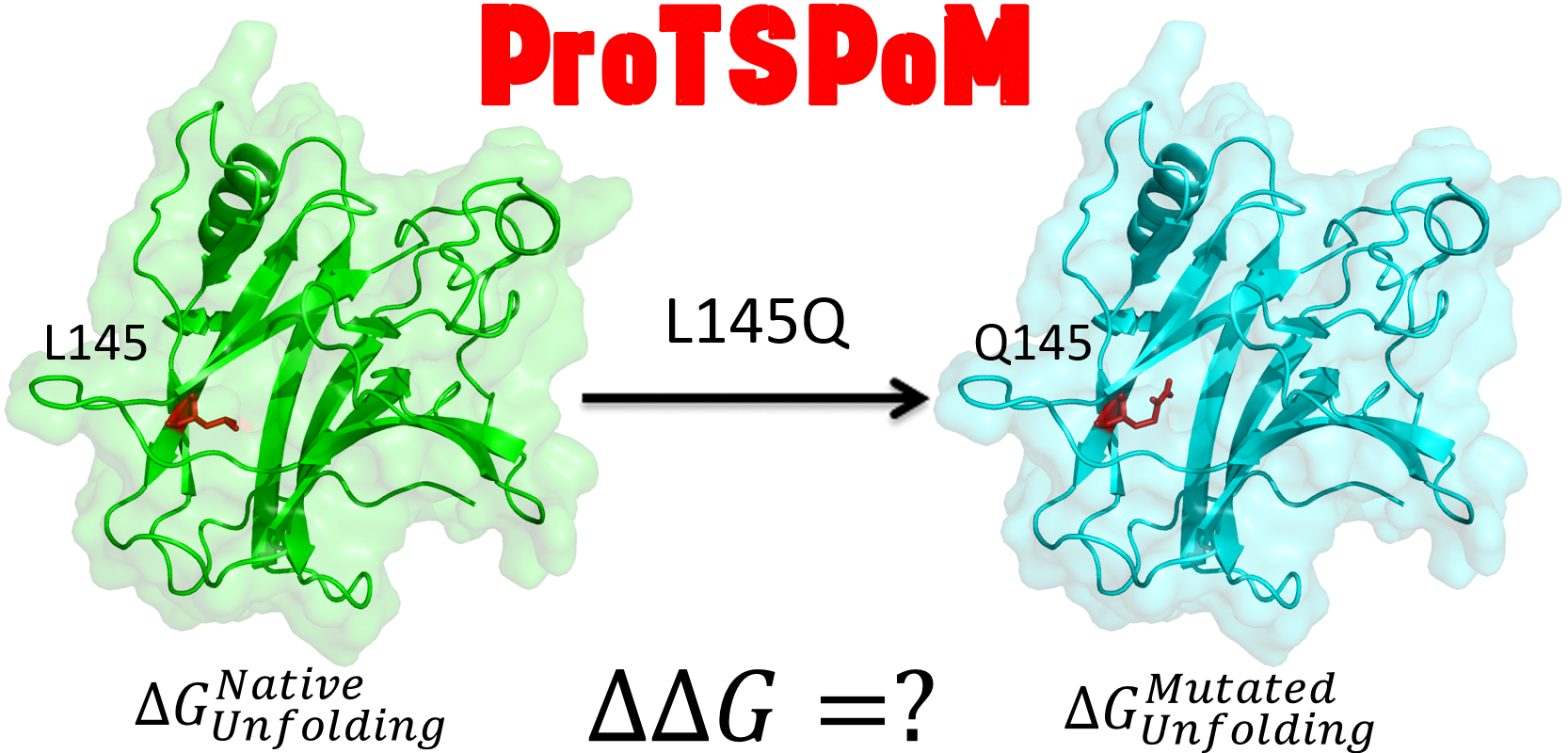
ProTSPoM uses a combination of Random Forest Regressors and Gradient Boosted Regressors along with residue properties, fold level attributes, environmental compatibility, and evolutionary information to predict the change in Gibbs free energy originating out of single point missense mutations. ProTSPoM outperforms all existing the methods in both the Pearson correlation coefficient and root-mean-squared-error parameters for the S2648, S350, S1925, and p53 databases. J Chem Inf Model 60(6):3315-3323
We introduce MaTPIP, a cutting-edge deep-learning framework for predicting PPI. MaTPIP stands out due to its innovative design, fusing pre-trained Protein Language Model (PLM)-based features with manually curated protein sequence attributes, emphasizing the part-whole relationship by incorporating two-dimensional granular part (amino-acid) level features and one-dimensional whole-level (protein) features. What sets MaTPIP apart is its ability to integrate these features across three different input terminals seamlessly. MatPIP also includes a distinctive configuration of Convolutional Neural Network (CNN) with Transformer components for concurrent utilization of CNN and sequential characteristics in each iteration and a one-dimensional to two-dimensional converter followed by a unified embedding. The statistical significance of this classifier is validated using McNemar's test. Comput Methods Programs Biomed 244:107955 (12 pages)
Protein phosphorylation is one of the essential post-translation modifications playing a vital role in the regulation of many fundamental cellular processes. We propose a LightGBM-based computational approach that uses evolutionary, geometric, sequence environment, and amino acid-specific features to decipher phosphate binding sites from a protein sequence. Proteins 88(2):284-291
Using biological insights, we construct an evolutionary profile to encode the amino acid variability in different positions of the target protein from its structural homologs. We propose an evolutionary profile guided replica-exchange Monte Carlo search algorithm that ensures faster convergence in protein design using a greedy strategy and confirms appreciable exploration and exploitation of the sequence-structure fitness surface. The simulation terminates dynamically after detective a stagnant situation. A series of sequence and structure level validations establish the goodness of our design. IEEE/ACM Trans Comput Biol Bioinform
Protein backbone alternation due to insertion/deletion or mutation operation often results in a change of fundamental biophysical properties of proteins. The proposed work intends to encode the protein stability changes associated with single-point deletions (SPDs) of amino acids in proteins. The encoding will help in the primary screening of detrimental backbone modifications before opting for expensive in vitro experimentations. J Proteome Res 18(3):1402-1410
NIP_NSc is a software to identify the normalized interface packing and normalized surface complementarity at the protein-protein contacts. The software has shows demonstrated performance in discriminating biological interfaces from non-biological ones. The time-efficient performance of the software allows the integration in the large scale screening of protein-protein contacts. FEBS Lett 584(6):1163-1168